暮色苍茫
生命就像一台精密的仪器,必须维持正常的工作状态—稳态。稳态,顾名思义,需要有一定的稳定性,这对于生命这样一个复杂系统,其实是不小的挑战。生命,对内要保证新陈代谢的持续性和恒定性,对外要形成对无数外界刺激的合适反应。在这样的动态环境中,生命允許稳态出现较小的、暂时的偏离,这种偏离过后,可以重新恢复到稳态。但是,内外部刺激形成的较大紊乱则会打破稳态平衡,令这台仪器无法正常工作,我们就生病了。
药物是对付疾病的最常用的手段,然而,研制药物是一件极其复杂、也非常奇妙的事情。让小小的药物分子帮助身体重建复杂生命系统的稳态,我们人类做到了,而且越做越好。
那么,药物是什么?它们是如何工作的?如何在成千上万的化学分子中找到能帮助我们的药物分子?
药物必须有“靶子”
谈到药物,就避不开靶点。任何药物都必须和体内特定目标(靶点)结合,对靶点介导的生物过程加以调节,才能达到治疗疾病的目的。因此,想了解药物,就必须先了解靶点。

在人体细胞中,有两类生物分子是常见的药物靶点:核酸和蛋白质。核酸就像公司的老板,平时主要工作是发号施令;
蛋白质是做具体工作的员工。因此,大部分药物的靶点都是细胞内的打工仔—蛋白质。
蛋白质种类繁多,其中有两类是药物研发的主要目标。
第一类药物目标蛋白质叫酶。人体就像一个大试管,时刻进行着各种各样的化学反应。消化食物、产生能量、传递信号、抵御病菌……任何生物过程都离不开化学反应。这些化学反应很给力,美中不足的是反应速度太慢,于是酶就来帮忙了。酶是催化剂,可以极大地提升这些反应的速度,最高可以提升几十万亿倍。人体内的酶种类繁多,如胃部的消化酶,如果缺了它们,食物就无法被快速分解吸收,人也就无法生存了。酶的另一个作用是让化学反应的速度变得可控,细胞可以通过改变酶的数量或者活性让反应快慢自如。因为酶的作用如此重要,它的活性变化可以轻易改变生物过程,酶也就成为药物的主要目标之一。
另一类常见的药物目标是被称为受体的蛋白质。和人一样,细胞也要互通信息,也会对外界环境做出反应。特定环境下会形成一类叫配基的分子,配基可以结合相应受体。配基/受体结合会引发一系列生物变化,影响体内很多生物过程。例如,胰岛素和胰岛素受体的结合控制着血糖水平,神经递质和其受体结合控制着人的心理活动和身体行为。因此,对配基/受体结合的调节自然也就成为了药物的主要目标。
药物VS身体
药物进入体内,会发生两件事,搞清楚这两件事,对药物的理解才算“功德圆满”。这两件事其实可以归纳为两个问题:药物对身体做了什么?身体又对药物做了什么?
研究“药物对身体做的事”有一个专门学科—药物效应动力学。既然被称为药物,当然要对身体做点儿事,最起码要对药物靶点起到预期的调节作用。药物通常还会有一些非特异性的脱靶现象,这是指结合之前没有预期的靶点,也会产生研究者不希望产生的生物效应。
研究“身体对药物做的事”也有一个专门学科—药物代谢动力学。药物必须在身体的作用位点达到一定的浓度并维持一定的时间才有效果。但是,药物是外源分子,身体有一整套天然的防御机制来修饰、降解和排泄外源分子。这些防御机制对于药物在体内的分布、代谢和滞留时间有很大影响。药物种类繁多,其物理化学性质各异,人体防御机制对不同药物产生的影响差异也很大,无法外推,必须通过实验加以研究。药物代谢动力学是药物研发中非常重要的一环,因为它决定了什么样的给药途径(口服、皮下注射、静脉注射等)和给药频率才能让药物在体内形成可以产生疗效的积累。
从化学“小分子”到生物学“大分子”
药物没有“隔空点穴”的功夫,想改变靶点的生物功能,就必须和靶点“亲密无间”地结合。靶点是分子,因此绝大部分药物也是分子级别的,可以分为小分子药物和大分子药物两大类。为什么这样划分?这就要从药物研发的“进化”过程谈起了。
药物研发和化学密不可分。绝大部分药物是地球上本来并不存在的化学分子,想打造出这些“前所未见”的分子,必须依靠化学这个“出神入化”的工具。即使是具有良好治疗效果的天然产物,想成为药物也往往需要化学方法的再加工和完善,直接拿来就能成药的天然产物非常罕见。
因此,化学学科的进步直接影响着药物的开发方法。从科技史上看,化学的发展先于生物学,因此早期的药物研发以化学方法为主。化学家在实验室合成的药物分子一般分子量为几百道尔顿(分子量单位),这种分子“身材娇小”,统称为“小分子药物”。小分子药物非常普遍,常见的抗生素、过敏药物、抗炎药物以及化疗药物都是小分子药物。
随后,生物学在药物研发领域快速跟进,特别是基因工程和蛋白质工程技术的发展,让开发者可以大规模地高效利用微生物来合成全新的蛋白质(即发酵),并使用这些蛋白质作为药物。蛋白质的分子量一般在几万到几十万道尔顿,这种分子“人高马大”,因此这类药物统称为“大分子药物”,也被称为“生物制剂”。
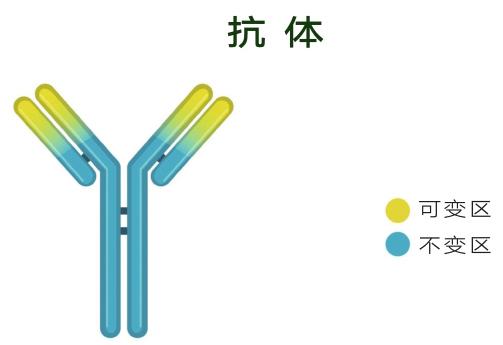
在大分子药物中,单克隆抗体(单抗)占据着主导地位。抗体也是蛋白质,但却是一种很特殊的蛋白质,因为每个抗体都能结合一个不同的抗原,即抗体的特异性。那么,什么物质可以作为抗原呢?答案是:没有限制,没有条件,种类繁多。所以,抗体具有多样性。抗体还有一个优点,不同的抗体共享一个非常相似的总体结构。这个结构像字母Y,只有Y的头上两个区域有变化,因为这两个区域负责结合不同抗原。由于抗体的结构相似性,研究者打造一个抗体研发平台,就可以合成不同的抗体,一“台”多用,非常高效。特异性、多样性和高效性让抗体成为药物科学家的好朋友。
不过,在大分子藥物研究领域,抗体不是唯一的选择。除了抗体,大分子药物还包括重组蛋白质药物和核酸药物等。
你也许觉得,小分子、大分子只是分子量大小不同而已,都是药物,何必区分?实际上,这两类药物从研发到后期生产都很不一样,需要不同的专长和知识背景。简单来说,小分子药物专家不一定会开发大分子药物,大分子药物专家也很难研发出小分子药物。所以,“小分子还是大分子”是一个关键问题,下面就聊聊这里面的“玄机”。
药物一般通过两个方法改变靶点功能,改变靶点的催化功能(酶)或者改变靶点和其他分子的相互结合。对于具有催化功能的酶来说,它们拥有一个“活性中心”,反应在此进行。这个“活性中心”常常具有小而深的特点,很适合“塞”一个小分子进去。因此,在改变酶的活性这方面,小分子药物得天独厚。但是和酶的“活性中心”相反,靶点和其他生物分子之间的作用面往往又大又浅,“浅”让小分子无处着手,“大”让小分子力不从心。因此,对于干涉分子之间相互作用,大分子药物更加有效。
细胞有一层脂性的细胞膜。大分子药物个头大,亲水基团多,因此无法穿越细胞膜,只能局限于细胞外部或者细胞膜表面朝外的靶点,对于细胞内部靶点鞭长莫及。例如,辉瑞的mRNA新冠疫苗,必须包裹在一层脂性“外衣”里才能穿膜而过。小分子药物则身材娇小,见缝插针,而且研究者可通过结构修饰来提升整个分子的亲脂性。因此,小分子药物可以横穿细胞膜,结合细胞内部靶点,极大地增加了小分子药物的靶向范围。
大分子药物是蛋白质或者核酸类生物分子,在经过胃部消化和肠道吸收后,这些分子会被变性降解而失去功能。因此,大分子药物无法口服,一般是通过静脉注射给药。小分子是全新的化学分子,体内肠胃尚没有进化出降解它们的机制,因此它们可以被身体完整地吸收,进入血液。所以,小分子药物不仅可以注射,还可以口服。如果医生给你开了一种口服药物,你可以自信地判断:“这是小分子药物。”
小分子药物又能进入细胞,又能口服,难道它毫无缺点?当然不是。小分子因为个头小,和靶点的结合位点也就小,在体内出现具有类似位点的其他生物分子的概率就大,再加上小分子无孔不入,就使得小分子药物更容易出现非特异性结合,产生所谓的脱靶效应。因此,小分子的毒副作用相对比较大。大分子则相反,和靶点的接触面大,而且只“看得见”细胞外部生物分子,因此较少脱靶,也就更加安全。
另外,抗体是身体的“熟人”,正常情况下我们身体中存在无数抗体,既然是“熟人”,身体就不会驱赶它,因此抗体在体内的滞留时间较长。例如,大分子单抗药物一般可以在体内存在几个星期,所以抗体的给药频率经常是每三周打一针。小分子是身体的“陌生人”,身体有各种机制“必先除之而后快”,导致小分子容易被身体快速清除,需要频繁给药才能维持有效浓度。
在生产、运输和储存方面,这两类药物也有很多差异。小分子的化学合成相对简单,而且精确可控。大分子结构复杂,合成中变量多,产品质量控制难度很大,但是这些缺点也使得生产大分子仿制药非常困难,相当于延长了制药公司的药物保护期,增加了经济效益。小分子一般相对稳定,便于储藏和运输。大分子不够稳定,需要低温保存,在存储和运输方面面临更大的挑战。
所以,关于“大分子还是小分子”的问题,要综合权衡各种因素,包括靶点的功能和位置、给药方法和频率、给药的便利程度、毒副作用的耐受程度、合成技术平台的成熟度以及运输、储存、销售、商业回报等,才能得到最佳答案。
研发药物的步骤
药物研发的第一步是找对靶点,这一步说起来容易做起来难。因为人体内的靶点成千上万,精确锁定靶点需要深厚的生物学和病理学专业知识作为基础。如果这一步错了,后面的研发就像在沙滩上盖楼,只会以悲剧收场。
现在,让我们以“寻找一个酶的小分子抑制剂”为例子,来看看药物研发的具体步骤。
擅长理性思维的科学家,首先想到的是研究目标酶上那个“活性中心”的形状,然后找一个形状相同的小分子放进去,就像拼图一样。这个想法是有道理的,而且也不断有人在尝试,但是成功率并不高。失败的原因有很多,比如,这个“活性中心”不是静态的,而我们现在还没有很好的办法预测其动态变化。另外,对于小分子和蛋白质之间的相互作用,研究者目前还没有做到可以量化的程度,仅仅形状的嵌合并不代表两者之间作用力的最大化。这些都影响了理性设计药物的潜力。
现在,最常用的方法是随机筛选小分子法。具体而言,就是首先建立一个随机的小分子库,里面有成千上万结构各异的小分子。然后,把每一个小分子和目标酶放在一起,通过一个分析方法来测量哪些小分子抑制了这个酶的活性。可能你会好奇,小分子库又是如何建成的? 其实,小分子库是在多年药物研发过程中,药物化学家们一点一点合成出来的,完全是科学家心血的结晶。目前,世界级大制药公司的小分子库一般都在百万这个数量级。想筛选这么多分子,必须采用“高通量”的分析方法,这种方法为随机筛选奠定了基础。
经过上面的海选,往往会找到成百上千个小分子可以抑制这个酶的活性,不过千万不能高兴得太早了,因为可能遇到“假阳性”。假阳性是指一个小分子看起来抑制了这个酶,其实并没有。原因多种多样,比如,是小分子里的杂质在影响酶的活性,或者是小分子形成聚合物导致酶的失活,也可能是分析方法的误差。实际上,初选得到的活性小分子中可能大多数都是假阳性,这些假阳性的小分子要经过一系列的分析手段逐步加以排除,这就是二级筛选。






