邢丽华


摘 要:本文旨在对泊沙康唑的合成工艺进行研究和改进。新工艺路线以4-(4-(4-硝基苯基)-1-哌嗪基)苯酚为起始原料,经缩合、还原、酰胺化、关环、水解等反应得到泊沙康唑。实验表明,本工艺路线操作简便,产品杂质少,反应条件温和,适合工业化生产。
关键词:泊沙康唑;合成;真菌药物;合成工艺
中图分类号:TQ465 文献标识码:A 文章编号:1673-260X(2021)08-0067-03
1 引言
泊沙康唑(posaconazole)是新一代由德国先灵葆雅公司研制的新型广谱抗真菌药,是第二代三唑类抗真菌药物,2005年10月20日被欧盟和美国FDA批准上市[1,2]。泊沙康唑化学名为4-[4-[4-[4-[[(3R,5R)-5-(2,4-二氟苯基)-5-(1,2,4-三唑-1-基甲基)氧杂戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基]-2-[(2S,3S)-2-羟基戊-3-基]-1,2,4-三唑-3-酮,是通过抑制真菌羊毛甾醇14α-去甲基化酶起作用[3]。泊沙康唑是亲脂的三唑类抗真菌药,抗菌谱较广,主要用于治疗由念珠菌属、隐球菌属引起的真菌血症,呼吸、消化道、尿路真菌病、腹膜炎和脑膜炎,经FDA批准泊沙康唑还可用于治疗对伊曲康唑和氟康唑耐药性较强的口咽念珠菌病[4-10],具有良好的安全性和耐受性。
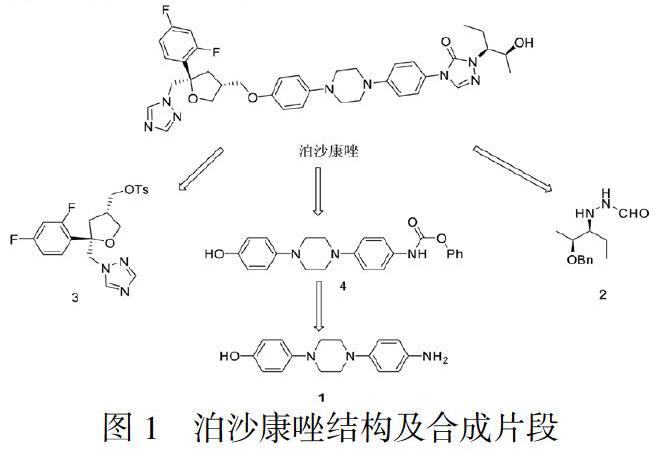
目前已报道泊沙康唑的合成工艺较多[11-16],但主要是围绕以下三个合成片段(片段1、2、3)开展的,具体结构见图1。
目前存在的问题是从化合物1到化合物4的制备过程中会生成新的杂质[17],即图2结构:
该杂质难以除去且在后续合成过程中会参与反应,生成新的杂质,造成杂质传递,影响后续中间体及目标产物泊沙康唑的质量。为了解决这个难题,本文设计了新的合成路线,见图3:以4-(4-(4-硝基苯基)-1-哌嗪基)苯酚为起始原料,首先与氯甲基甲醚反应生成中间体6,保护羟基;硝基再经钯碳还原得到氨基得到中间体7,接着与氯甲酸苄酯反应生成中间体8,与外购化合物2反应得到中间体9,在酸性条件下脱去羟基上保护基得到中间体10,与外购化合物3发生缩合反应得到中间体11,最后脱去羟基上保护基苄基得到目标产物泊沙康唑。新设计的工艺路线避免产生难以除去的杂质,提高了泊沙康唑的产品质量,且工艺操作简单、工艺条件温和,易于实现工业化生产。
2 实验部分
2.1 主要仪器和试剂
仪器:AV-400MHz型核磁共振仪;高效液相色谱LC-20AT(日本岛津公司);DF-101S集热式恒温加热磁力搅拌器(郑州予华)。
原料:4-(4-(4-硝基苯基)-1-哌嗪基)苯酚(百灵威,含量≥97%);氯甲基甲醚(北京偶合科技有限公司,≥98%);氯甲酸苯酯(邦成化工,≥98%);2-[(1S,2S)-1-乙基-2-苄氧基丙基]肼甲醛(天津卡普希科技有限公司,≥98%);(5R-CIS)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1H-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(西安瑞联新材料股份有限公司,≥97%)。
2.2 实验过程
2.2.1 1-(4-(甲氧基甲氧基)苯基)-4-(4-硝基苯基)哌嗪(中间体6)制备
向一个干燥的3L的三口瓶中加入4-(4-(4-硝基苯基)-1-哌嗪基)苯酚(66.0g,0.22mol),然后加入四氢呋喃(620mL)。冷却降温至-5~0℃,搅拌10分钟,分批加入叔丁醇钾(49.4g,0.44mol),控温搅拌1小时;慢慢滴加氯甲基甲醚(21.2g,0.26mol),2小时内滴加完毕,继续控温-5~0℃继续搅拌1小时,再缓慢升至室温继续搅拌3小时,采用HPLC监控反应至完全。反应完毕,体系缓慢倒入3L水中,析出固体,室温搅拌1小时,抽滤,用500mL纯化水洗涤固体至滤液呈中性,40℃鼓风干燥得到黄色中间体6(58.4g,77.1%)。1H-NMR(400MHz,CDCl3):3.22-3.25(m,4H),3.48(s,3H),3.56-3.59(m,4H),5.13(s,2H),6.86-6.85(m,2H),6.89-6.93(m,2H),7.00-7.02(m,2H),8.13-8.16(m,2H)。
2.2.2 1-(4-(甲氧基甲氧基)苯基)-4-(4-硝基苯基)哌嗪-4-(4-(4-(甲氧基甲氧基)苯基)哌嗪-1-基)苯胺(中间体7)制备
将四氢呋喃(310ml)、中间体6(21.2g,0.0617mol)加入到500ml三口瓶中,加入10%钯碳(1.24g),室温通入氢气反应24小时。HPLC跟踪反应至中间体6消失即反应结束。反应液用硅藻土抽滤,再用THF(250ml)淋洗滤饼,滤液直接进行下一步反应,无需进行纯化。取5ml滤液直接浓缩得到固体送分析。1H-NMR(400MHz,CDCl3):3.17-3.19(m,4H),3.22-3.25(m,4H),3.44-3.48(m,5H),5.12(s,2H),6.66-6.68(m,2H),6.85-6.87(m,2H),6.91-6.94(m,2H),6.97-7.00(m,2H)。
2.2.3 苯基(4-(4-(4-(甲氧基甲氧基)苯基)哌嗪-1-基)苯基)氨基甲酸酯(中间体8)制备
将上步反应液降温至0-10℃,缓慢滴加氯甲酸苯酯(12.0g,0.0768mol),滴加30min。滴加完畢自然升至室温,继续反应6小时。HPLC跟踪反应至化合物7含量小于0.5%即可进行后处理。向体系中加入纯化水200ml,析出固体,继续搅拌1小时。抽滤,滤饼依次经50ml纯化水、50ml乙醇、50ml正己烷洗涤,得到灰白色固体中间体8(23.2g,87.1%)。
1H-NMR(400MHz,CDCl3):3.16-3.21(m,8H),3.33-3.36(m,3H),5.09(s,2H),6.94-6.91(m,6H),7.19-7.26(m,3H),7.36-7.44(m,4H),9.98(s,1H)。
2.2.4 2-(2S,3S)-2-(苄氧基)戊烷-3-基)-4-(4-(甲氧基甲氧基)苯基)哌嗪-1-基)苯基)-2,4-二氢-3H-1,2,4-三唑-3-酮(中间体9)制备
向1.0L三口瓶中加入1,2-二甲氧基乙烷(150mL)、中间体8(9.97g,0.023mol)、化合物2(8.27g,0.035mol)、三乙胺(15g,0.148mol),加毕,搅拌,升温至80℃反应10-12h,TLC跟踪中间体8消失,反应完毕。加入500mL纯化水,搅拌0.5小时,分液,有机相40℃减压浓缩得到粗品,再加入甲基叔丁基醚450ml重结晶,得到土黄色固体中间体9(10.9g,85.0%)。
1H-NMR(400MHz,CDCl3):0.86-0.87(m,1H),1.11(m,3H),1.62(m,1H),3.33(m,3H),3.57-3.60(m,9H), 3.92(m,1H),4.63(s,1H),6.02(s,2H),6.65-6.73(m,4H),6.90(m,2H),7.09(m,2H),7.31(s,4H),7.94(s,1H)。
2.2.5 2-((2S,3S)-2-(苄氧基)戊烷-3-基)-4-(4-(4-羟基苯基)哌嗪-1-基)苯基)-2,4-二氢-3H-1,2,4-三唑-3-酮(中间体10)制备
向500mL三口瓶中依次加入中间体9(10.0g,0.0179mol)、乙醇(150mL)、1N盐酸(20mL)室温搅拌反应3小时。TLC跟踪中间体9小事,反应完毕。加入1N氢氧化钠溶液调节pH=6-7,加入二氯甲烷(200mL*2)萃取,分液。有机相分别用饱和碳酸钠溶液(50mL)和饱和盐水(50ml)洗涤,有机相40℃减压浓缩得到土黄色粗品。将固体加入300ml甲基叔丁基醚进行重结晶,得到类白色固体即中间体10(7.71g,83.9%)。
1H-NMR(400MHz,CDCl3):0.86-0.87(m,1H),1.12(m,3H),1.62(m,1H),3.57-3.61(m,9H),3.92(m,1H), 4.63(s,1H),6.61(m,4H),6.90(m,2H),7.09(m,2H),7.31(s,4H),7.94(s,1H)。
2.2.6 (4-(4-(4-(5-(2,4-二氟苯基)-5-(1,2,4)三唑-1-基甲基-氧杂戊-3-基甲基)-苯基)-哌嗪-1-基)-苯基)-氨基甲酸苄酯(中间体11)制备
氮气保护下,向250ml三口瓶中加入DMSO(70 mL)、化合物3(7.07g,0.0158mol)、中间体10(7.00g, 0.0137mol),室温搅拌0.5小时,固体全部溶解。加入氢氧化钠和水的混合溶液(0.9g氢氧化钠/3.3mL水配制),室温搅拌0.5小时。体系升温至40℃,反应8-12小时。TLC跟踪至中间体10消失,反应完毕。加入3N盐酸调节pH=5-6,搅拌0.5小时。缓慢向体系中滴加水(50mL),析出大量白色固体,滴加完毕继续搅拌1.0小时。抽滤,滤饼用水(20ml)淋洗,40℃鼓风干燥得到类白色固体即中间体11(8.9g,86.2%)。
2.2.7 4-[4-[4-[4-[[(3R,5R)-5-(2,4-二氟苯基)-5-(1,2,4-三唑-1-基甲基)氧雜戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基]-2-[(2S,3S)-2-羟基戊-3-基]-1,2,4-三唑-3-酮(泊沙康唑)合成
在500mL三口烧瓶中加入中间体11(8.0g, 0.010mol)、甲醇(200mL)、5NHCl(25mL)、10%钯碳(0.96g),氮气置换3次,室温通入氮气反应。TLC跟踪反应至中间体11消失。反应完毕,抽滤,滤液用2N氢氧化钠调节pH=10,体系析出类白色固体。固体加入甲醇100mL中加热至完全溶解,加入活性炭(0.8g),搅拌30分钟脱色。趁热抽滤,滤饼用热的甲醇20ml淋洗。滤液冷却析晶,抽滤得到白色固体即泊沙康唑(6.50g,91.7%)。
1H-NMR(400MHz,CDCl3):8.33(s,1H),7.78(s,1H),7.50-7.54(s,1H),7.23-7.30(m,3H),7.08-7.11(s,2H),6.92-7.01(m,8H),6.78-6.81(m,2H),4.67- 4.69(d,1H),4.57—4.58(d1H),3.99-4.04(m,1H),3.66-3.84(m,4H),3.29-3.34(m,8H),3.15-3.16(m,2H),2.12-2.16(m,2H),1.68-1.72(m,1H),1.11-1.13(m,3H),0.71-0.76(m,3H)。
3 结论
由于泊沙康唑合成路线复杂、反应条件苛刻、合成路线过长、大量使用昂贵的催化剂、污染环境、不利于大规模工业生产等缺点,目前国内没有泊沙康唑原料药备案和制剂获批厂家,市场上所有的泊沙康唑制剂均为进口,因药品价格昂贵,给国内的用药患者带来巨大的经济负担。
通过研究,一方面,实现了泊沙康唑国产化,降低了国内患者的用药成本,同时也缓解了临床用药需求。另一方面,泊沙康唑新的合成工艺起始原料廉价易得,反应收率高,减少传统工艺难以除去的杂质,降低了成本,易于实现工业化。
参考文献:
〔1〕Julia J Chang,Alejandro Villar-Prados,MD,et al.Concurrent Pseudohyperaldosteronism and primary glucocorticoid deficiency from Posaconazole[J].Journal of Endocrine Society,2021,5(01):124-125.
〔2〕Van Daele Ruth,Brüggemann Roger J,Dreesen Erwin, et al. Pharmacokinetics and target attainment of intravenous posaconazole in critically ill patients during extracorporeal membrane oxygenation[J]. Journal of Antimicrobial Chemotherapy, 2021,76(05):1234-1241.
〔3〕Epps Quovadis J.,Epps Kevin L.,Zobell Jeffery T., et al. Optimization of antimicrobials in the treatment of cystic fibrosis pulmonary exacerbations:?II. Therapies for allergic bronchopulmonary aspergillosis[J].Pediatric Pulmonology,2020,55(12):3541-3572.
〔4〕Lambrix Arathi A,Swanson Hope D,Pauley Jennifer L,et al. Experience using intravenous posaconazole in paediatric and young adult oncology patients[J]. Journal of Antimicrobial Chemotherapy,2020,75(12):3682-3687.
〔5〕Van Daele Ruth, Spriet Isabel, Maertens Johan. Posaconazole in prophylaxis and treatment of invasive fungal infections:
a pharmacokinetic, pharmacodynamic and clinical evaluation[J]. Expert Opinion on Drug Metabolism & Toxicology, 2020,16(07):539-550.
〔6〕Salehi Mohammadreza, Shahi Farhad, Rizvi Fatema Sadaat, et al. Combination antifungal therapy without craniotomy in an immunocompromised patient with rhino-orbito-cerebral mucormycosis:
A case report[J].Caspian Journal of Internal Medicine, 2020,11(02):227-230.
〔7〕刘洁,胡小平,刘伟.三唑类抗真菌新药泊沙康唑和伏立康唑简介[J].菌物学报,2018,37(10):1391-1398.
〔8〕L.Tang, X.-F. Yang, M. Qiao, L. Zhang, X.-W, et al. Posaconazole vs. voriconazole in the prevention of invasive fungal diseases in patients with haematological malignancies:
A retrospective study[J]. Journal de Mycologie Médicale, 2018,28(2):379-383.
〔9〕Santiago Grau, Rafael Cámara, Manuel, et al.Cost-effectiveness of posaconazole tablets versus fluconazole as prophylaxis for invasive fungal diseases in patients with graft-versus-host disease after allogeneic hematopoietic stem cell transplantation[J]. The European Journal of Health Economics,2018,19(04):627-636.
〔10〕徐宵寒,張路,段明辉.评价泊沙康唑预防血液系统疾病患者粒细胞缺乏期侵袭性真菌病的效果[J].中国感染控制杂志,2017,16(01):32-35.
〔11〕郭彦飞,蓸卫,包玉盛,等.泊沙康唑(API)生产化工艺研究及杂质谱分析[J].山东化工,2018,47(23):47-55.
〔12〕闫新创,陈平,贺敏,等.泊沙康唑重要中间体的合成与表征[J].广州化工,2013,41(10):107-109.
〔13〕黄松.一种泊沙康唑中间体的合成方法[P].CN201710204742A.
〔14〕安敏,王伟,邓洪丽,邓.泊沙康唑关键中间体合成的新方法[J].2019,40(02):31-35.
〔15〕靳凤民,张静.泊沙康唑的合成[J].中国新药杂质,2015,24(05):560-564.
〔16〕李驰,王欣.泊沙康唑合成路线图[J].中国药物化学杂志,2020,30(03):182-187.
〔17〕杨祥龙,李金凤,邵伟,等.泊沙康唑合成工艺研究进展[J].化工时刊,2019,33(11):22-37.
猜你喜欢 合成 酰胺型含氮硼酸酯的合成中国化工贸易·中旬刊(2020年6期)2020-12-28浅析高分子材料合成中有机化学的应用分析科学与财富(2020年24期)2020-10-27奥美沙坦酯合成新方法研究中国医药导报(2019年7期)2019-05-13金丝桃素药理作用以及制备方法研究概况中国民族民间医药·上半月(2018年8期)2018-09-182,6—二氟苄酰胺类生物碱的合成及抑菌活性研究安徽农业科学(2018年25期)2018-05-14新型异胞嘧啶类物质的生物活性研究卷宗(2017年36期)2018-03-09利伐沙班的合成工艺研究科学与财富(2017年17期)2017-06-16关于有机化学在高分子材料合成中应用情况的研究卷宗(2017年6期)2017-06-06羟基蛋氨酸钴配合物的合成、表征与热分析湖南师范大学学报·自然科学版(2017年1期)2017-03-14雷迪帕韦及其关键中间体的合成研究上海医药(2016年9期)2016-06-03




