彭凌 林锦铨 李玲慧 刘博婷 蔡巩林


摘要:
本研究测定猪丹毒丝菌临床毒株SG7的全基因组序列,并运用生物信息学方法对测定的全基因组序列及猪丹毒丝菌表面保护性抗原A基因(SpaA)进行分析。SG7菌株的基因组全长为1 834 291.00 bp,G+C含量为36.3%,基因总数1 846个。将SG7菌株与GenBank中8条完整的猪丹毒丝菌全基因组序列进行比较,发现国内外不同菌株间基因组的基本信息存在不同程度差异。基于全基因组单核苷酸多态性(SNPs)的系统发育分析结果表明,9株菌株聚为3个分支,国内菌株并不完全处于同一分支,SG7菌株与国内常见菌株不属于同一进化分支。SpaA基因高变区的分析结果显示,9株菌株亦可分为3个SpaA型,SG7菌株为携带Met203的高致病性菌株。除SG7菌株外,其他8株菌株的SpaA基因分型结果与基于全基因组SNPs分析的结果一致,说明SG7菌株的遗传背景复杂。本研究结果可为猪丹毒丝菌基因组整体水平研究和疫苗的研发奠定基础。
关键词:
猪丹毒丝菌;全基因组测序;表面保护性抗原A(SpaA)
中图分类号:
S852.61 文献标识码:
A 文章编号:
1000-4440(2021)03-0694-05
Whole-genome sequencing and SpaA gene analysis of a Erysipelothrix rhusiopahiae strain
PENG Ling, LIN Jin-quan, LI Ling-hui, LIU Bo-ting, CAI Gong-lin
(Henry Fok College of Biology and Agriculture, Shaoguan University, Shaoguan 512005, China)
Abstract:
In this study, the whole genome sequence of Erysipelothrix rhusiopahiae clinical strain SG7 was determined, and the whole genome sequence and the surface protective antigen A (SpaA)gene were analyzed using bioinformatics method. The full length of SG7 genome was 1 834 291.00 bp, and the total quantity of the predicted genes were 1 846 with a G+C content of 36.3%. Different degrees of differences of genomic basic information between different strains were found by comparing the complete genome sequences of SG7 and eight E. rhusiopahiae found in GenBank. Phylogenetic analysis based on genome-wide single nucleotide polymorphisms (SNPs) revealed that nine strains could be clustered into three distinct clades, and strains isolated from China were not in the same clade, while SG7 and common strains isolated from China did not belong to the same clade. The analysis results of hypervariable region of SpaA showed that nine strains could also be divided into three SpaA types, and SG7 was a highly pathogenic strain carrying Met203. Except for SG7, the SpaA genotyping results of other eight strains were consistent with the analytic results obtained based on the genome-wide SNPs analysis, which indicated that the genetic background of SG7 was complex. The results can provide data basis for the research at the whole genome level and development of E. rhusiopahiae vaccine.
Key words:
Erysipelothrix rhusiopahiae;whole-genome sequencing;surface protective antigen A (SpaA)
豬丹毒丝菌(Erysipelothrix rhusiopahiae)又称红斑丹毒丝菌,属于丹毒丝菌属(Erysipelothrix),呈纤细杆状,是一种革兰氏阳性的人畜共患病病原菌。猪丹毒是由猪丹毒丝菌引发的一种急性、热性传染病,能导致家禽、畜类的一系列皮肤感染及败血症。人类因外伤局部感染猪丹毒丝菌,会发生类丹毒,感染时手指出现斑点或肿起[1-2]。在20世纪80至90年代,猪丹毒与猪肺疫、猪瘟并称中国养猪业三大传染病,给养猪业带来巨大的损失并影响人类健康[3]。近年来,猪丹毒在中国多个省份均有发生,且发病率呈上升趋势[4]。
目前预防和控制猪丹毒疫病的主要手段是接种疫苗,传统疫苗虽然对猪丹毒的预防起到一定作用,但仍有保护率低、保护周期短等缺点。细菌的毒力因子是人们研制新型疫苗的标靶[5]。有研究结果表明,猪丹毒丝菌表面保护性抗原A(SpaA)是其重要的毒力因子[6-7],可以通过介导猪丹毒丝菌与猪内皮细胞的黏附而侵入宿主发挥作用[7]。同时亦有很多研究结果证实了SpaA蛋白的N端区域为免疫保护区,对猪丹毒丝菌毒株有良好的免疫保护作用[8-10]。全基因组测序是通过DNA测序仪对物种基因进行测序的一种生物信息学手段,全基因组测序分辨率高、分析速度快,为传染性疾病的调查提供了新的方法,对疫病监测具有重要的指导意义[11]。本研究拟通过测定分离自广东省韶关市的猪丹毒丝菌毒力菌株SG7[12]的全基因序列,利用相关软件进行全基因组的注释分析,并分析SpaA的遗传多样性,以期为猪丹毒丝菌的后续研究提供有利的数据支撑,也为新型亚单位疫苗的开发和改良奠定理论基础。
1 材料与方法
1.1 菌株
从广东省韶关市某猪丹毒发病猪场分离获得猪丹毒丝菌菌株SG7,通过动物试验鉴定为毒力菌株,半致死量(LD50)为7.3×103CFU[12]。
1.2 基因组的测序与注释
将得到的猪丹毒丝菌DNA样品进行Illumina HiseqTM测序,获得原始图像数据,再经过碱基识别得到原始测序数据。但鉴于Illumina测序过程中产生的错误会对最终的分析结果造成影响,为了数据的准确性,通过软件FastQC对测序数据质量进行可视化评估,再利用软件Trimmomatic[13]过滤原始数据,以保证信息分析的准确性。使用软件SPAdes[14]对得到的测序数据进行拼接,然后对拼接得到的重叠群进行填补空隙。在修正拼接过程中,产生剪辑错误及小片段插入缺失会影响分析的准确性,因此需要采用软件PrInSeS-G[15]进行序列矫正,得到最终的结果。拼接结果采用软件Prokka[16]进行基因元件的分析预测,同时利用软件RepeatModeler鉴定SG7基因组上的重复序列。最后,采用软件NCBI Blast+将蛋白质氨基酸序列与COG、KEGG、VFDB等数据库的数据进行比对,获得蛋白质功能注释信息。
1.3 SpaA基因碱基序列分析
从测定的SG7菌株以及GenBank上公布的猪丹毒丝菌全基因组序列中提取SpaA基因碱基序列,并翻译为氨基酸序列,根据前人描述的方法[17-18]分析SpaA基因432 bp高变区对应蛋白质N端的氨基酸序列。
1.4 SG7菌株与其他猪丹毒丝菌的比较基因组学分析
对SG7菌株与GenBank上公布的猪丹毒丝菌完整的全基因组序列进行比较基因组学分析,并构建基于全基因组单核苷酸多态性(SNPs)系统进化树图。
2 结果与分析
2.1 猪丹毒丝菌菌株SG7基因组的基本特征
表1显示,猪丹毒丝菌菌株SG7的基因组全长为1 834 291.00 bp,G+C含量为36.30%。组分分析发现,基因总数为1 846个,碱基数为1 685 442.00个,基因平均长度为913.02 bp,占基因组全长的91.89%,蛋白质预测数為1 755个。采用软件Aragorn预测tRNA,预测数为55个,采用软件RNAmmer预测rRNA,预测数为3个。利用软件RepeatModeler对重复序列进行预测,SG7菌株共有107个平均长度为170.30 bp的重复序列,其中DNA重复元件有19个,低复杂度序列有12个,微卫星DNA序列有76个。
2.2 猪丹毒丝菌菌株SG7全基因组的注释
在猪丹毒丝菌菌株SG7已经预测的1 755个基因中,有1 316个基因注释到COG数据库中[19],占预测基因的74.99%,将这些基因信息依据COG分类标准进行分类,可分为19类;有1 079个基因注释到KEGG数据库中[20],占预测基因的61.48%,可将这些基因划分为24类代谢通路。
VFDB[21]是专门用于研究致病因子的数据库,包括SetA和SetB 2部分。将猪丹毒丝菌菌株SG7预测得到的基因碱基序列、蛋白质氨基酸序列与VFDB数据库中的数据进行比对,把基因和其相对应的毒力因子功能注释信息结合起来,有108个基因注释到SetA数据库,有114个基因注释到SetB数据库。
CARD[22]是耐药基因数据库,能为药物作用研究以及环境治理提供研究依据。将猪丹毒丝菌菌株SG7预测得到的基因碱基序列、蛋白质氨基酸序列与CARD数据库中的数据进行比对,有23个基因注释到CARD数据库。
PHI-base[23]是病原与宿主互作数据库,用于寻找药物干预的靶基因。将猪丹毒丝菌菌株SG7预测得到的基因碱基序列、蛋白质氨基酸序列与PHI-base数据库中的数据进行比对,有34个基因注释到PHI-base数据库。
2.3 猪丹毒丝菌SpaA基因分析
当前GenBank上公布的猪丹毒丝菌完整基因组序列共有8条,从SG7和GenBank中的8株猪丹毒丝菌的基因组序列中提取SpaA基因432 bp高变区,并对相应蛋白质的N端进行分析,结果表明,9个菌株中有5个菌株为Met203/Ile257型,3个菌株为Ile203/Ile257型,1个菌株为Ile203/Leu257型。
2.4 SG7菌株与其他猪丹毒丝菌全基因组的比较分析
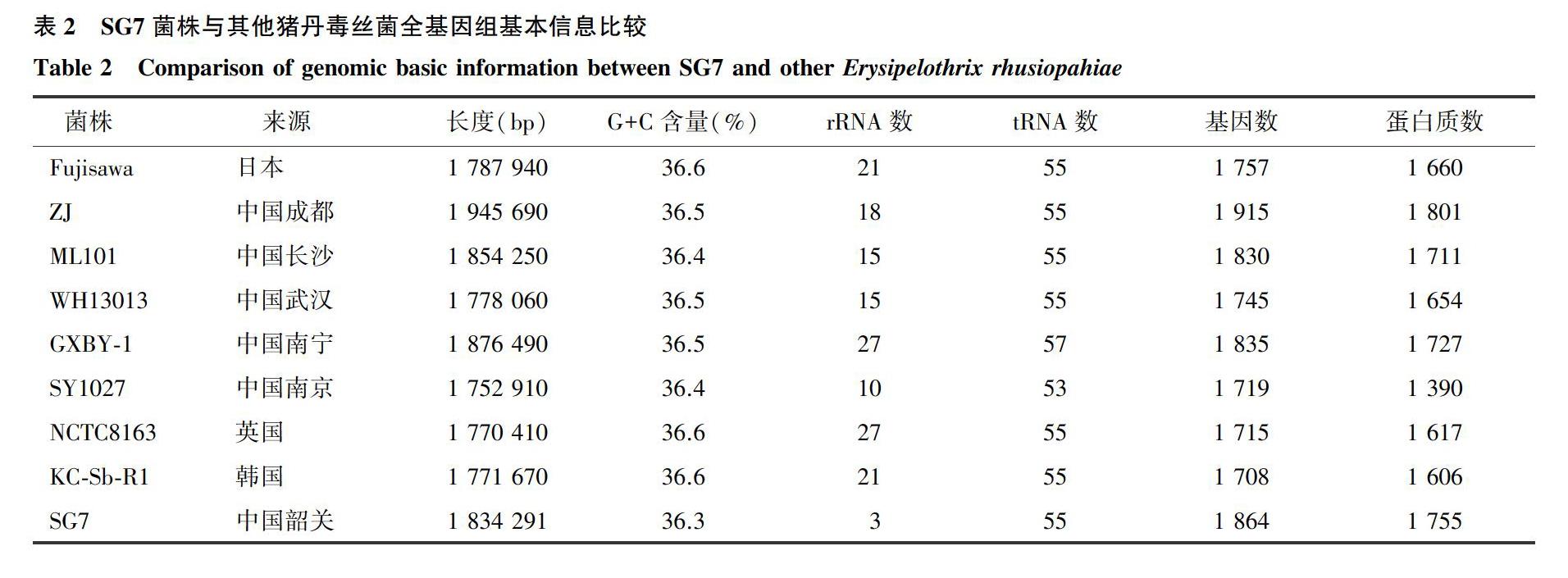
将SG7菌株与GenBank中8株猪丹毒丝菌菌株的全基因组基本信息进行比较,结果(表2)显示,猪丹毒丝菌全基因组长度为1 752 910~1 945 690 bp,G+C含量维持在36.3%~36.6%,基因数保持在1 708~1 915个,而蛋白质数为1 390~1 801个,表现出较大差异;tRNA保持在55个左右,但不同菌株中的rRNA数量差异较大,SG7的rRNA仅为3个,数量远低于其他菌株。9个菌株的共有基因为1 234个,特有基因为193个。
将上述8株菌株与SG7菌株的全基因组进行比较分析,并构建系统进化树图(图1),该进化树含有3个分支,SG7菌株与英国分离株NCTC8163、韩国分离株KC-Sb-R1、中国南京分离株SY1027同处一个分支;而其他4个国内分离株处于第二分支;日本分离株Fujisawa单独为一个分支。
3 讨论
猪丹毒作为一种人畜共患病,给养猪业带来巨大的经济损失并影响人类健康,该病的主要预防手段是疫苗接种,但传统灭活疫苗存在免疫效果不持久,不能产生细胞免疫等局限性[24]。猪丹毒的药物治疗大多采用β-内酰胺类抗生素,但过分依赖抗生素,会导致耐药性的出现以及药物残留的风险[25-27],有地区就曾分离出猪丹毒丝菌青霉素耐药菌株[28]。因此,开发更为有效的亚单位疫苗是科学防控猪丹毒的关键。尽管从1876年首次分离该菌至今已经过去了100多年,但我们对猪丹毒丝菌分子机制的了解仍然十分有限[29-30]。2011年德国北部暴发大肠杆菌疫情,造成数千人感染,数十人死亡,但病原菌株的病原培养特性和细菌多位点序列分型(MLST)分析结果均与肠出血性大肠杆菌菌株相差甚远,有学者怀疑是新型的致病性大肠杆菌,经过全基因组测序分析发现,该菌株与肠聚集性大肠杆菌的亲缘关系更为接近,在进化上处于同一分支,但在进化过程中获得了志贺毒素编码基因stx2及多种耐药基因,使其获得了更强的生存能力,造成了病原菌的广泛传播[31]。可见,全基因组分析对病原菌株的监测以及遗传变异检测有很大帮助,全基因组测序为我们从分子水平研究猪丹毒丝菌提供了便利。
本研究测定了猪丹毒丝菌菌株SG7临床毒株[12]全基因组序列,并通过使用相关的生物信息学软件及数据库对其进行注释,揭示了该菌株的基因组结构特征,丰富了猪丹毒丝菌的基因组数据库。将SG7与GenBank上公布的8株猪丹毒丝菌完整基因组序列的基本信息进行比较,除tRNA数和G+C含量比较一致外,基因组长度、基因数、蛋白质数、rRNA数SG7菌株与国内外其他菌株存在不同程度的差异,这也许与不同地区饲养管理以及环境因素等存在差异有关。基于全基因组SNPs的系统发育分析结果表明,9株菌株聚为3个分支,这与Forde等[32]、Ogawa等[18]的研究结果一致。国内菌株并不完全处于同一分支,SG7菌株与南京分离株SY1027、英国分离株NCTC8163、韩国分离株KC-Sb-R1为同一个分支,而南宁分离株GXBY-1、成都分离株ZJ、武汉分离株WH13013、长沙分离株ML101这4个国内分离株则为另一分支,说明国内菌株至少有2个不同的进化途径,SG7菌株与英国分离株NCTC8163关系最近。SpaA高变区的氨基酸序列分析结果表明,9株菌株可分为3个SpaA型,SG7菌株和南宁分离株GXBY-1、成都分离株ZJ、武汉分离株WH13013、长沙分离株ML101均为Met203/Ile257型。Uchiyama等[17]认为携带Met203的分离株为高致病性菌株。Ogawa等[18]对分离自日本的34株菌株进行分析,结果表明这些菌株的SpaA基因分型结果与基于全基因组SNPs分析获得的分类结果是一致的,本研究中除SG7菌株外,其他8个菌株亦如此。与SG7菌株处同一分支的其他菌株均为Ile203/Ile257型,SG7菌株为携带Met203的高致病性菌株(LD50为7.3×103CFU),但它与携带Met203的其他菌株不处于同一进化分支,因此推测SG7菌株在进化过程中也许与携带Met203的进化分支的菌株接触后进行了SpaA基因的重组交换。本研究结果为猪丹毒丝菌基因组整体水平研究和亚单位疫苗的开发提供了数据基础。
参考文献:
[1] GORBY G L, PEACOCK J E. Erysipelothrix rhusiopathiae endocarditis:
microbiologic, epidemiologic, and clinical features of an occupational disease[J]. Reviews of Infectious Diseases, 1988, 10(2):
317-325.
[2] WANG Q, CHANG B J, RILEY T V. Erysipelothrix rhusiopathiae [J].Veterinary Microbiology, 2010, 140(3/4):405-417.
[3] 周緒斌,丹 尼,李 聪,等. 猪丹毒——古老的传染病是否从中国规模化猪场消失了?[J].今日养猪业,2009(5):22-24.
[4] 李文春,杨仕标,李富祥. 一株猪丹毒杆菌的分离鉴定及SpaA基因序列分析[J].上海畜牧兽医通讯,2020(3):9-12.
[5] 周作勇,李和贤,杨浩钺,等. 伪结核棒状杆菌毒力因子的研究进展[J].中国人兽共患病学报, 2017,33(12):1115-1119.
[6] BORRATHYBAY E, GONG F J, ZHANG L, et al. Role of surface protective antigen A in the pathogenesis of Erysipelothrix rhusiopathiae strain C43065[J]. Journal of Microbiology and Biotechnology, 2015, 25(2):206-216.
[7] HARADA T, OGAWA Y, EGUCHI M, et al. Phosphorylcholine and SpaA, a choline-binding protein, are involved in the adherence of Erysipelothrix rhusiopathiae to porcine endothelial cells, but this adherence is not mediated by the PAF receptor[J]. Veterinary Microbiology, 2014, 172 (1/2):216-222.
[8] 吾鲁木汗·那孜尔别克,张 磊,何 翠,等. 猪丹毒丝菌天然SpaA和重组SpaA-N免疫保护效果的评价[J].微生物学报, 2010,50(3):367-372.
[9] 姚焱彬,陆 萍,杨志鹏,等. 猪丹毒丝菌SpaA基因原核表达及表达蛋白质的免疫原性分析[J].畜牧兽医学报,2017,48(3):492-500.
[10]蒋志琼,钟泽民,谭博敏,等. 丹毒丝菌SpaA基因免疫保护区的克隆及其在毕赤酵母中的表达[J].华南农业大学学报,2015,36(3):20-25.
[11]周江林,彭小川,胡明达,等. 基于全基因组测序的细菌进化研究进展[J].生物技术通讯,2018, 29(6):844-850.
[12]周 迪,杨旭夫,彭 凌. 红斑丹毒丝菌的分离鉴定及药敏试验[J].动物医学进展,2020,41(2):33-38.
[13]BOLGER A M, LOHSE M, USADEL B. Trimmomatic:
a flexible trimmer for Illumina sequence data[J]. Bioinformatics, 2014,30(15):2114-2120.
[14]BANKEVICH A, NURK S, ANTIPOV D, et al. SPAdes:
a new genome assembly algorithm and its applications to single-cell sequencing[J]. Journal of Computational Biology, 2012,19(5):455-477.
[15]MASSOURAS A, HENS K, GUBELMANN C, et al. Primer-initiated sequence synthesis to detect and assemble structural variants[J]. Nature Methods, 2010,7(7):485-486.
[16]SEEMANN T. Prokka:
rapid prokaryotic genome annotation[J]. Bioinformatics, 2014,30(14):2068-2069.
[17]UCHIYAMA M, YAMAMOTO K, OCHIAI M, et al. Prevalence of Met-203 type SpaA variant in Erysipelothrix rhusiopathiae isolates and the efficacy of swine erysipelas vaccines in Japan[J].Biologicals,2014,42(2):109-113.
[18]OGAWA Y, SHIRAIWA K, OGURA Y, et al. Clonal lineages of Erysipelothrix rhusiopathiae responsible for acute swine erysipelas in Japan identified by using genome-wide single nucleotide polymorphism analysis [J]. Applied & Environmental Microbiology, 2017, 83(11):
e00130-17.
[19]TATUSOV R L, GALPERIN M Y, NATALE D A, et al. The COG database:
a tool for genome-scale analysis of protein functions and evolution[J]. Nucleic Acids Research,2000, 28(1):33-36.
[20]KANEHISA M, GOTO S. KEGG:
kyoto encyclopedia of genes and genomes[J]. Nucleic Acids Research, 2000,28 (1):27-30.
[21]CHEN L, ZHENG D, LIU B, et al. VFDB 2016:
hierarchical and refined dataset for big data analysis-10 years on[J]. Nucleic Acids Research, 2016, 44:D694-D697.
[22]MCARTHUR A G, WAGLECHNER N, NIZAM F, et al. The comprehensive antibiotic resistance database[J]. Antimicrobial Agents and Chemotherapy, 2013, 57(7):3348-3357.
[23]URBAN M, PANT R, RAGHUNATH A, et al. The pathogen-host interactions database (PHI-base):
additions and future developments[J]. Nucleic Acids Research, 2015, 43:D645-D655.
[24]姚焱彬. 豬丹毒丝菌灭活疫苗的制备及其对小鼠的免疫效力研究[D].合肥:安徽农业大学,2018.
[25]张 珍,施开创,王孝德,等. 2015-2017年广西鸡源沙门氏菌耐药性与致病性的相关性分析[J].南方农业学报,2019,50(10):2350-2358.
[26]徐重新,刘 敏,张 霄,等. 农药危害风险及其残留检测用广谱特异性抗体研究进展[J] .江苏农业学报,2019,35(2):489-496.
[27]王志芳,雷 燕,肖 俊,等. 广西罗非鱼主产区养殖池塘抗生素残留状况分析[J].南方农业学报,2019,50(4):891-897.
[28]胡晓芬,王 静,熊剑锋,等. 一起猪丹毒杆菌病例的临床分离鉴定及诊治[J].养猪,2014(2):107-109.
[29]王力波. 猪丹毒丝菌广西分离株生物学特性研究及其基因组生物信息学分析[D].长春:吉林农业大学,2015.
[30]曹文尧. 丹毒丝菌表面保护抗原A作为重组亚单位疫苗和核酸疫苗的初步研究[D].乌鲁木齐:新疆大学,2007.
[31]CUI Y, LI D, YANG R. Shiga toxin-producing Escherichia coli O104∶H4:an emerging important pathogen in food safety[J]. Chinese Science Bulletin, 2013, 58(14):1625-1631.
[32]FORDE T, BIEK R, ZADOKS R, et al. Genomic analysis of the multi-host pathogen Erysipelohrix rhusiopathiae reveals extensive recombination as well as the existence of three generalist clades with wide geographic distribution[J]. BMC Genomics, 2016, 461(17):1-15.
(责任编辑:王 妮)





